4.3 Lab Exercises
Overview
This lab will walk us through the process of annotating a genome for repeats.
We will do two major things in this lab:
Learn how to run EDTA, an all-in-one transposon annotation pipeline
Build an insertion time distribution for LTR elements of major superfamilies
“Be for real, don’t be a stranger” - Spice Girls
Task A
Now that we have a fully phased genome assembly, we can begin the process of annotation. There are two major phases to annotation: repetitive elements, and genes. First, we need to annotate repetitive elements so that we can mask them before annotating genes, effectively “hiding” the repeats from gene annotation and prediction programs. Repeats are the most frustrating aspect of annotating a genome. Not only are they ubiquitous, and typically different between every species, if you do a poor job of repeat annotation, your gene annotation will also be poor. Remember: repetitive elements contain genes!
EDTA is an all-in-one repeat annotation pipeline. It runs a handful of programs that identify different kinds of repetitive elements, such as LTR_FINDER, LTRharvest, Generic Repeat Finder, and HelitronScanner. Then it filters these annotations to build a custom repeat library, and creates a .gff file of repeat annotations for the genome assembly. Here’s an overview of the pipeline:
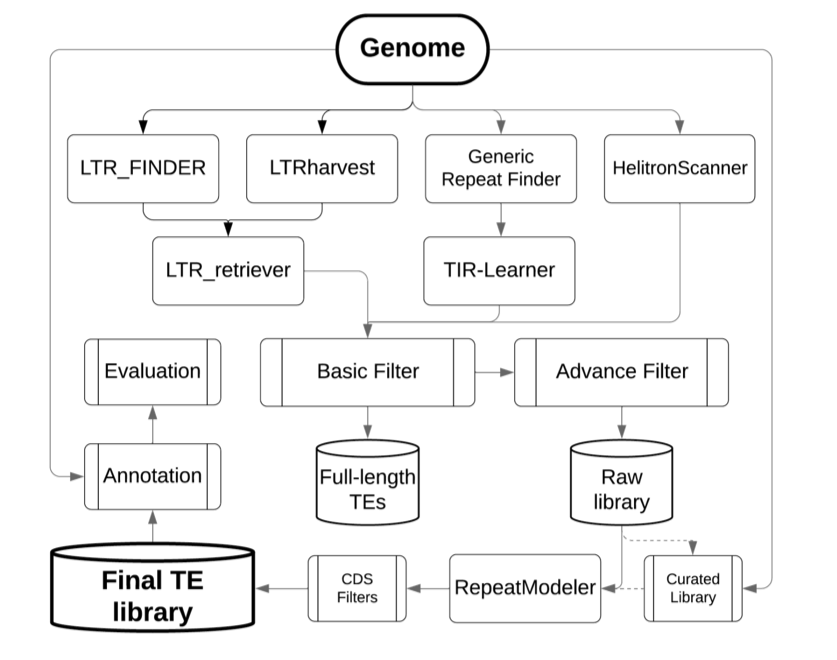
Image Source: EDTA GitHub Page
{kind=link}
The EDTA workflow First, read the github wiki page, then create a new Conda environment for EDTA:
conda create -n EDTA
conda activate EDTA
Then install via Conda.
conda install -c bioconda -c conda-forge edta
…. This is taking forever, isn’t it? My install just hangs. I think it finishes in a few hours, but we don’t have that kind of time to waste, do we? Sometimes Conda installs are just painfully slow.
Mamba was developed to beat this problem. Mamba is a reimplementation of the conda package manager in C++. It allows for:
parallel downloading of repository data and package files using multi-threading
libsolv for much faster dependency solving, a state of the art library used in the RPM package manager of Red Hat, Fedora and OpenSUSE
core parts of mamba are implemented in C++ for maximum efficiency
Use mamba to accelerate the installation:
conda install -c conda-forge mamba
mamba install -c conda-forge -c bioconda edta python=3.6 tensorflow=1.14 'h5py<3'
Test your installation by typing:
EDTA.pl -h
Task B
First, read some of the caveats of EDTA. It looks like all we need, at minimum, is just a genome file .fasta file. We have two, one for each haplotype, so we will divide-and-conquer as a class. We’ll decide which half of the class takes haplotype1 and which half takes haplotype2 when we meet.
It does mention that “make sure sequence names are short and simple”. Okay, I’ve taken care of this, and renamed our .fasta headers to be “Qv1_chr1”, “Qv1_chr2”, etc.
Now, read the EDTA Usage section, and see if you can launch a full run on your own. Remember that you can use at most 4 threads.
Mastering Content
Next, we want to produce some figures that describe the repeat landscape in the genome. In particular, we want to show the percent sequence divergence of similar repeats, in order to describe any “bursts” of repeat expansion.
We’re not the first people using EDTA who want to do the same. Check out this Github Issues describing the same thing: https://github.com/oushujun/EDTA/issues/92
If you’ve used R before, this should look somewhat familiar. Open the R terminal in your PRAXIS welcome page, install the required packages (google if you don’t remember how.